El Laboratorio de Enfermedades Metabólicas (LEM), perteneciente al Instituto de Nutrición y Tecnología de los Alimentos (INTA), Doctor Fernando Monckeberg Barros de la Universidad de Chile, forma parte de una unidad especializada en la pesquisa, diagnóstico y seguimiento de pacientes con Errores Innatos del Metabolismo.
El Laboratorio existe hace más de 30 años y fue calificado como Centro de Referencia por el Ministerio de Salud en 1999 en las “Normas para el óptimo desarrollo de Programas de Búsqueda Masiva de Fenilquetonuria, Hipotiroidismo Congénito y otros Errores Innatos del Metabolismo”.
Los errores innatos del metabolismo son enfermedades monogénicas y en su mayoría de herencia autosómica recesiva. La alteración de un gen produce un defecto enzimático, que conduce a alteraciones bioquímicas características de cada enfermedad metabólica. La mayoría de estas patologías se manifiestan en la edad pediátrica, desde las primeras horas de vida hasta la adolescencia, con signos y síntomas inespecíficos y que son similares a otras patologías; no reconocerlas conlleva a graves secuelas, siendo las más frecuentes: convulsiones, desnutrición y retardo mental. La prevención de estas secuelas con un diagnóstico oportuno es el gran desafío de los pediatras. Con el objetivo de prevenir las secuelas nuestro laboratorio tiene a disposición de los equipos de salud una serie de ensayos para la medición de metabolitos y actividades enzimáticas que buscan llegar al diagnóstico de estas patologías. Además de entregar a cada paciente y su familia una asistencia de calidad junto al cariño de cada uno de nosotros en este esfuerzo diario.
Misión: Diagnóstico de pacientes con EIM, ocupando técnicas de laboratorio cualitativas y cuantitativas.
Visión: Entregar resultados de análisis altamente confiables, por lo que el aseguramiento de la calidad de nuestro trabajo es uno de nuestros pilares.
Los EIM pueden ser pesquisados en forma preventiva en los primeros días de edad, con la finalidad de llegar en forma temprana al diagnóstico y con esto evitar secuelas de la progresión de la enfermedad. A esta estrategia se le denomina Pesquisa Neonatal, la cual puede abarcar 1 o más patologías, lo que está definido por el análisis que se aplique en un país o en un laboratorio.
Lo más habitual en países o regiones donde no se aplica un Programa de Pesquisa Neonatal o este abarca un número reducido de patologías, es que la estrategia diagnóstica sea reactiva, es decir cuando un médico al observar la sintomatología del paciente solicite exámenes que confirmen la sospecha clínica.
Nuestro Laboratorio está en permanente desarrollo, para dar respuesta a ambas estrategias diagnósticas. Por esto es que nos hemos posicionado como referentes en Chile, recepcionando muestras de todas las regiones del país, para este tipo de análisis. Así también tenemos vínculo con MINSAL en el soporte del tratamiento nutricional de algunas de estas patologías.
En este contexto, nuestro laboratorio permite dar soporte al estudio de patologías como:
La Pesquisa Neonatal para detección de EIM y otras patologías nace en la década de 1960, con la pesquisa de Fenilquetonuria (PKU). Esta estrategia implica la aplicación de un análisis de laboratorio a muestras de la población de recién nacidos de una región. El área de Pesquisa Neonatal (PN) del Laboratorio de Enfermedades Metabólicas existe hace más de 20 años y fue clave para que en 1992 el Ministerio de Salud (MINSAL) comenzará el Programa Nacional de Búsqueda Masiva de Fenilquetonuria e Hipotiroidismo Congénito. Esto llevo a que en 1999 fuésemos calificados como Centro de Referencia por el MINSAL para el desarrollo y seguimiento del Programa Nacional de Búsqueda Masiva de Fenilquetonuria (PKU). La aplicación del Programa permite mejorar la calidad de vida de los niños chilenos con PKU, al prevenir la discapacidad que esta enfermedad produce. Este programa otorga cobertura al 100% de los nacidos vivos en todos los servicios públicos a nivel nacional y tiene como objetivo prevenir el retardo mental si es detectada y tratada en el periodo neonatal.
Además, el área de PN ofrece servicios de screening neonatal los cuales consiste en analizar una muestra de sangre en papel filtro que es tomada desde las 40 horas de vida del recién nacido, esta muestra es tomada en el hospital o clínica donde nació su hija o hijo, y será enviada a nuestro laboratorio para su análisis, contamos con la pesquisa de las siguientes patologías;
1. Fenilquetonuria (PKU): 1.1 Forma clásica de PKU
1.2 Hiperfenilalaninemias
2. Hipotiroidismo Congénito.
3. Enfermedad de orina con olor a jarabe de arce (MSUD).
Equipamiento:
Contamos con un Fluorimetro Infinite 200 PRO de Labsystems.
Nuestra área además, dentro de sus principales funciones realiza la monitorización constante de Fenilalanina de los pacientes ya diagnosticados con Feniquetonuria e hiperfenilalaninemia, ayudando en su tratamiento continuo ya que con esto se adecúa la ingesta de macro y micronutrientes de acuerdo a su evolución física y fisiopatológica.
La Pesquisa Neonatal para detección de EIM y otras patologías nace en la década de 1960, con la pesquisa de Fenilquetonuria (PKU). Esta estrategia implica la aplicación de un análisis de laboratorio a muestras de la población de recién nacidos de una región. Tras la PKU se fueron agregando otras patologías, en donde un ensayo permitía la detección de una patología. En la década de 1990 se introduce la Espectrometría de Masas en Tándem (MSMS), que permite el análisis de múltiples metabolitos, permitiendo la detección de diversas patologías en un único ensayo. A este análisis se le denomina Pesquisa Neonatal Ampliada (PNA). Algunos países suman a esta PNA otras patologías, no posibles de detectar por MSMS, por lo que al hablar de PNA debemos definir en qué país o región se aplica, para poder saber el numeró de patologías incluidas en el análisis de laboratorio.
En 2002, nuestro laboratorio implemento el uso de la MSMS, como método de análisis, lo que nos permite el estudio de Aminoacidopatías, Acidurias Orgánicas y Defectos de B-Oxidación. En 2007 implementamos otras técnicas de Pesquisa Neonatal, lo que nos permitió ofrecer la PNA, que hacemos hasta la fecha y la ofrecemos como un examen que puede ser realizado a los recién nacidos de nuestro país.
Equipamiento: Contamos con Espectrómetro de Masas en Tándem TMAPI de Micromass, Espectrómetro de Masas en Tándem 3500 de ABSCIEX, asociado a HPLC Shimadzu, Fluorimetro Victor 2D de Perkin Elmer.
Detalle de Patologías incluidas en PNA de INTA
Patologías en detección (actualizado a marzo 2025)
Aminoacidopatías:
1- Aciduria Argininosuccinica
2- Citrulinemia Tipo 1
3- Enfermedad de orina con olor a jarabe de arce (MSUD)
4- Fenilquetonuria (PKU) + Hiperfenilalaninemias
5- Tirosinemia Tipo 1
6- Argininemia
7- Homocistinuria
Acidurias Orgánicas:
8- Deficiencia de 3-Hidroxi-3-Metilglutaril-CoA Liase (HMG)
9- Acidemia Glutárica Tipo I (AG-I)
10- Acidemia Isovalérica (AIV)
11- Deficiencia 3-Metilcrotonil-CoA Carboxilasa (Deficiencia de 3 MCC)
12- Acidemia Metilmalónica (AMM)
13- Deficiencia de Cofactor (Cbl A, B, C, D)
14- Acidemia Propiónica (AP)
15- Deficiencia Mitocondrial de Acetoacetil-CoA Tiolasa (Def. 3-Ketotiolase)
16- Deficiencia de Múltiples Carboxilasas Defectos de B-Oxidación
17- Deficiencia Neonatal de Carnitina Palmitoil Transferasa –Tipo I y II (CPT I; CPT-II)
18- Deficiencia 3-Hydroxi-Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena larga (LCHAD)
19- Deficiencia de Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena mediana (MCAD)
20- Deficiencia de Proteína Trifuncional (Deficiencia TFP)
21- Deficiencia de Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena muy largas (VLCAD)
DETECTADAS POR OTRAS TÉCNICAS:
22. Hiperplasia Adrenal Congénita.
23. Fibrosis Quística.
24. Hipotiroidismo Congénito.
25. Déficit de Biotinidasa.
26. Galactosemia Clásica.
Patologías secundarias a detectar (1) (actualizado a marzo 2025)
– Hipermetioninemia
– Deficiencia Isobutiril-CoA deshidrogenasa (IBDD)
– Deficiencia 2-Metilbutiril-CoA Deshidrogenasa (2MBD)
– Deficiencia 3-Metilglutaconil-CoA Hidratasa
– Deficiencia Carnitina/Acilcarnitinas Translocasa (CAT)
– Deficiencia Múltiple de Acil-CoA Deshidrogenasa (MADD o Acidemia Glutárica-Tipo II)
– Deficiencia de Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena corta (SCAD)
– Deficiencia de 3-Hydroxi-Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena corta (SCHAD)
– Deficiencia de Carbamoilfosfato Sintetasa (Def CPS)
– Síndrome de Hiperamonemia, Hiperornitinemia, Homocitrulinemia (HHH)
– Aciduria Malónica
– Deficiencia de 2,4-Dienoil-CoA Reductasa
– Deficiencia de Galactokinasa
– Deficiencia de Galactosa-4-Epimerasa
– Tirosinemia tipo 2 y tipo 3
– Citrulinemia tipo 2
– Deficiencia primaria de Carnitina (OCTN2)
(1) Patologías secundarias que nuestro laboratorio detecta pero que han presentado falsos negativos en programas de pesquisa internacionales.
Las enfermedades de depósito lisosomal son diversas y sólo para algunas existen Programas de Pesquisa Neonatal acotados y aun con resultados que ponen en duda su utilidad. Por esto es que, nuestro laboratorio ha desarrollado su experiencia, en técnicas que permitan al médico contar con análisis confirmatorios para el diagnóstico de algunas de estas patologías.
Se pueden clasificar en Mucopolisacaridosis, Esfingolipidosis, Mucolipidosis y Oligosacaridosis, lo que está dado solo por el tipo de metabolito que se acumula en los lisosomas.
Para Mucopolisacaridosis y Oligosacáridosis el laboratorio cuenta con métodos de aproximación al diagnóstico.
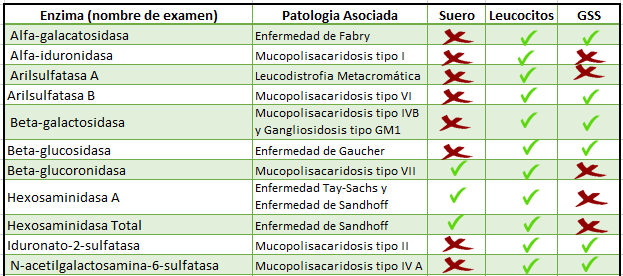
El Laboratorio de Enfermedades Lisosomales está en constante desarrollo de las pruebas diagnósticas, que corresponden a cuantificar la actividad enzimática asociada a una patología lisosomal específica. Estas determinaciones realizadas en suero o leucocitos aislados de muestras de sangre del paciente corresponden a los análisis “gold standard” para el diagnóstico de estas patologías.
Para algunas patologías es posible realizar los ensayos enzimáticos en muestras de sangre impregnadas en papel filtro (Whatman 903) denominadas “GSS”, sin embargo, estos tests solo deben ser considerados como pruebas de aproximación.
Debido a nuestro desarrollo, somos el único laboratorio en Chile nombrado por el MINSAL como proveedor para la confirmación diagnóstica de las patologías incluidas en la Ley 20.850, conocida como Ley Ricarte Soto (https://www.minsal.cl/leyricarte/). Las patologías incluidas en la Ley son: MPS I, MPS II, MPS VI, Enfermedad de Gaucher y Enfermedad de Fabry. Para acceder a las garantías de la Ley el médico a cargo del paciente debe inscribir al paciente en la plataforma de FONASA.
Equipamiento: Contamos con Espectrofluorimetro, Spectramax M2 de Molecular Devices.
Detalle de Patologías Lisosomales en nuestro laboratorio:
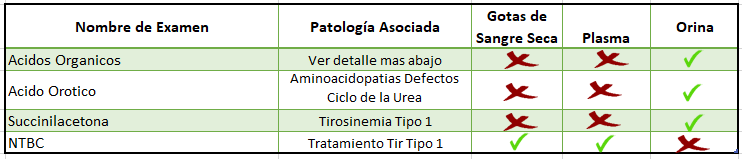
El Área de Cromatografía Gaseosa se implementó en nuestro laboratorio el año 2015, dando respuesta a la necesidad de dar diagnóstico a los casos de Acidurias Orgánicas (AO) en Chile. Hasta ese momento el diagnóstico solo era posible enviando muestras al extranjero. Las moléculas de bajo tamaño excretadas por la orina pueden ser detectadas mediante el uso de un cromatógrafo gaseoso acoplado a la espectrometría de masas. El diagnóstico de una AO se realiza estudiando caso a caso el patrón de excreción de los ácidos orgánicos en una muestra de orina. Anormalidades en la excreción en uno o más metabolitos pueden ser indicadores de una AO. El análisis de estos patrones es de carácter interpretativo y cualitativo. Al encontrar un patrón anormal, el informe indicará cuales son los metabolitos que se encuentran alterados y se comentará si son concordantes con alguna Aciduria orgánica.
Para una correcta interpretación, es importante contar con información clínica y nutricional por lo que solicitamos enviar el “Formulario de solicitud de examen de ácidos orgánicos” junto con la muestra.
A través de estos años hemos logrado incluir la cuantificación de algunos metabolitos (Succinilacetona y ácido Orótico). La cuantificación se realiza mediante método de dilución isotópica que permite la determinación de sus niveles en forma cuantitativa y confiable, el valor que se entrega se encuentra normalizado con los niveles de creatinina presente en la orina. Un caso particular es el diagnóstico de Tirosinemia Tipo 1 tras la cuantificación de Succinilacetona en orina. Esta patología esta incluida en la ley Ricarte Soto y somos el único prestador acreditado por el MINSAL a la fecha.
Análisis que realizamos en esta área son:
Equipamiento: Contamos con un GC-MS (7890A-5965C) Agilent . También con Sciex Triplequad 3500 (LC-MS/MS)
Detalle de patologías pesquisadas por test de Ácidos Orgánicos urinarios:
Aminoacidopatías
Aminoácidos de Cadena Ramificada
Acidurias Orgánicas
Desórdenes del Ciclo de Urea
Desórdenes del Metabolismo de la Pirimidina
Desórdenes del Metabolismo Energético Mitocondrial
Otros Desórdenes
Cuando un cuerpo clínico se enfrenta a un paciente con una Enfermedad Metabólica, es posible que los signos cardinales no sean tan evidentes, dificultando la sospecha hacia una patología específica. En estos casos el apoyo de tests de laboratorios ayudan en esta orientación y finalmente pueden establecer un diagnóstico. Por lo anterior, es que nuestro laboratorio está en constante desarrollo de técnicas de laboratorio, que permitan al cuerpo clínico aproximarse al diagnóstico definitivo.
Ante esta situación el examen de primera línea es el Perfil de Aminoácidos y Acilcarnitinas (PAC), que se realiza por espectrometría de Masas en Tándem. Este análisis permite, en un único ensayo, la cuantificación de más de 30 metabolitos, lo que permite sospechar o diagnosticar EIM asociados a Aminoacidopatías, Acidurias Orgánicas y Defecto de B-Oxidación.
Detalle de Patologías incluidas en PAC de INTA:
Aminoacidopatías:
1- Aciduria Argininosuccinica
2- Citrulinemia Tipo 1
3- Enfermedad de orina con olor a jarabe de arce (MSUD)
4- Fenilquetonuria (PKU) + Hiperfenilalaninemias
5- Tirosinemia Tipo 1
6- Argininemia
7- Homocistinuria
Acidurias Orgánicas:
8- Deficiencia de 3-Hidroxi-3-Metilglutaril-CoA Liase (HMG)
9- Acidemia Glutárica Tipo I (AG-I)
10- Acidemia Isovalérica (AIV)
11 Deficiencia 3-Metilcrotonil-CoA Carboxilasa (Deficiencia de 3 MCC)
12- Acidemia Metilmalónica (AMM)
13- Deficiencia de Cofactor (Cbl A, B, C, D)
14- Acidemia Propiónica (AP)
15- Deficiencia Mitocondrial de Acetoacetil-CoA Tiolasa (Def. 3-Ketotiolase)
16- Deficiencia de Múltiples Carboxilasas Defectos de B-Oxidación
17- Deficiencia Neonatal de Carnitina Palmitoil Transferasa –Tipo I y II (CPT I; CPT-II)
18- Deficiencia 3-Hydroxi-Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena larga (LCHAD)
19- Deficiencia de Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena mediana (MCAD)
20- Deficiencia de Proteína Trifuncional (Deficiencia TFP)
21- Deficiencia de Acil-CoA Deshidrogenasa de Ácidos Grasos de cadena muy largas (VLCAD).
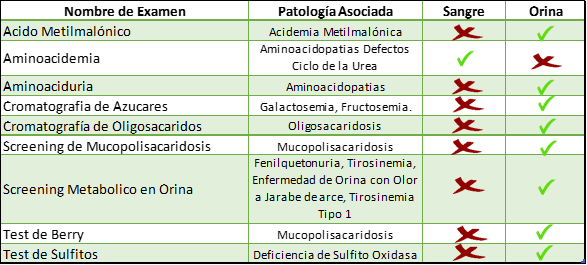
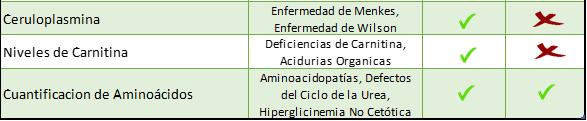
Si la sospecha diagnóstica es acotada a Defectos del Metabolismo Energético, a una entidad más específica como Mucopolisacaridosis, Desordenes de Azucares, Metabolismo del Cobre u otras, ofrecemos una serie de tests que pueden ser solicitados.
Dentro de esta gama de exámenes que ponemos a disposición para el estudio de EIM, podemos clasificarlos en Exámenes Cualitativos y Cuantitativos.
Dentro de los exámenes cualitativos están:
Dentro de los exámenes Cuantitativos están:
Equipamiento: Contamos con Espectrómetro de Masas en Tándem TMAPI de Micromass, Cromatógrafo Líquido asociado a Espectrómetro de Masas en Tándem 3500de ABSCIEX , Fluorímetro Perkin Elmer Victor 2D, Espetrofluorímetro SpectramaxM2 de MolecularDevices.
Listado de examenes disponible en nuestro laboratorio
Jefe de Unidad/Director Técnico
Debes hacer tu pedido por correo siempre indicando: Nombre completo, RUT y
edad del paciente. Nombre completo de quien retira, RUT del destinatario, lugar de recepción
(domicilio o sucursal Starken) y teléfono de contacto.
MN-TM-001 Manual de toma de Muestras. V17
AX-TM-002 Indicaciones Orina 24 hrs
AX-TM-003 Indicaciones Test Allopurinol
AX-TM-004 Indicaciones Gotas de Sangre Seca
AX-TM-005 Imágenes de muestras de GSS válidas y no válidas para análisis
AX-TM-006 Formulario de solicitud de exámenes
AX-TM-007 Formulario de solicitud de exámenes AO
**Este formulario debe ser enviado obligatoriamente cuando solicitan ácidos orgánicos.
AX-TM-010 Indicaciones para derivación técnicas lisosomales

Ingresa tu búsqueda y presiona [enter] o el botón "Buscar"